Collinearity Analysis
Collinearity
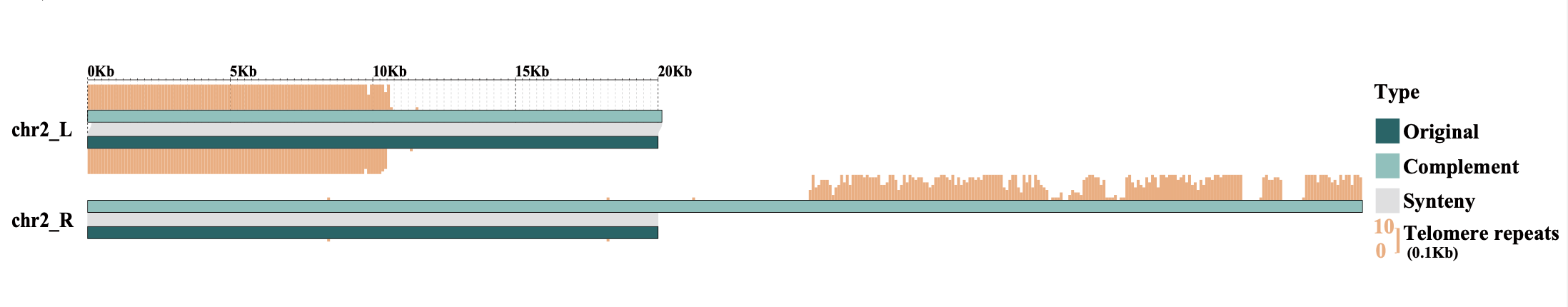
In collinearity analysis, the left and right ends of chromosomes from the original genome and the complemented genome (new_genome.fasta) are extracted for comparison, with the results output to genomeSyn_result. The number of telomeres at the corresponding chromosome ends of both genomes is also reported, including the files telomere.original.num.info and telomere.complement.num.info.
# optional arguments:
# -h, --help show this help message and exit
# -G1 , --genome1 Input the original genome file (FASTA format)
# -G2 , --genome2 Input new genome file (FASTA format)
# -pos The position information of telomere installed on the original
# genome
# -m , --motif Telomeric repeats sequences, e.g., plant: CCCTAAA(TTTAGGG),
# animal: TTAGGG(CCCTAA), etc.
# --bin The bins for calculating the number of telomeres are divided into.
# The bin size can be determined by the user, and the default value
# is 100.
# --genomeSyn1 GenomeSyn1 is in the order of original genome first and new genome
# second
# --genomeSyn2 genomeSyn2 is in the order of original genome second and new
# genome first
# --length Extract genome length
# -tel_max The maximum value of telomere frequency within the calculated
# length
# -C1 , --genome_color1 Set the color parameters of genome 1, the default is #04686b, or
# select other hexadecimal or RGB color codes, such as
# "#04686b"/"rgb(4,104,107)"
# -C2 , --genome_color2 Set the color parameters of genome 2, the default is #87c9c3, or
# select other hexadecimal or RGB color codes, such as
# "#87c9c3"/"rgb(135,201,195)"
# -C3 , --synteny_color Set the color parameters of the synteny blocks, the default is
# #DFDFE1, or select other hexadecimal or RGB color codes, such as
# "#DFDFE1"/"rgb(223,223,225)"
# -C4 , --telo_color Set the telomere color parameters for displaying genomes, the
# default is #fcaf7c, or select other hexadecimal or RGB color
# codes, such as "#fcaf7c"/"rgb(252,175,124)"
# -t , --threads Number of threads to use (default: 20)
$ telocomp_Collinearity -G1 original_geonme.fasta \
-G2 new_genome.fasta \
-pos telomere_positions.txt \
--length 20000 \
-tel_max 10 \
-m CCCTAAA --genomeSyn2
Collinearity result
Collinear alignment between the Morus notabilis original genome and the telomeric complemented genome (only chromosomes with complementary telomeres are shown).