Complement Module
Complement
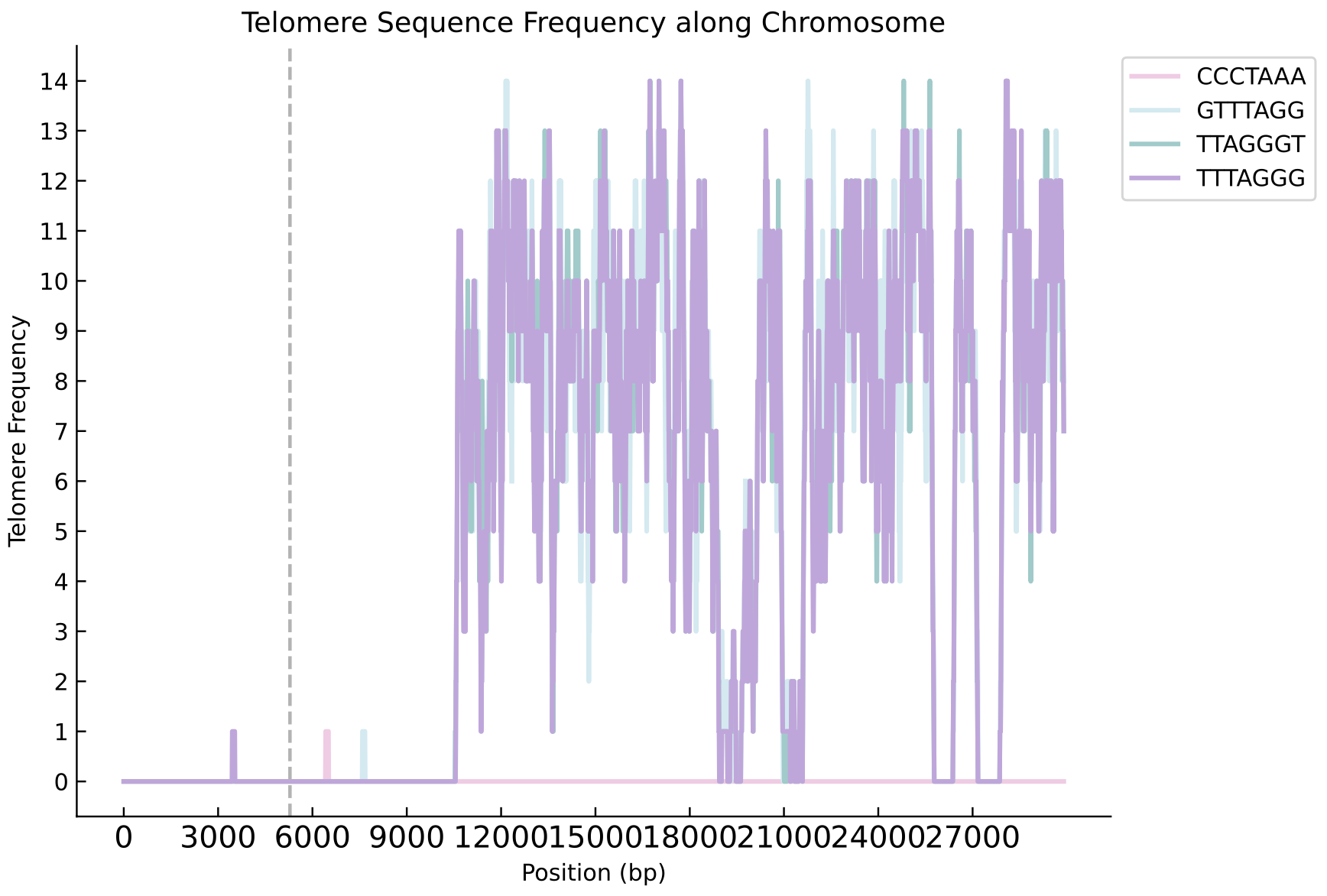
The Complement module complements the original genome with the assembled and polished reads, producing the completed genome file (new_genome.fasta).It also outputs the telomere position file (telomere_positions.txt), the telomere type file (telomere_repeats_info.txt), and density distribution plots of telomeres at the ends of each chromosome (stored in the telomere_plots18 folder).
# -h, --help show this help message and exit
# -G , --genome Input genome file (FASTA format)
# --dir_contigs Input polished contigs
# --dir_trim_L Input the trimmed reads. If the conditions are not met, extract the
# shortest reads.
# --dir_trim_R Input the trimmed reads. If the conditions are not met, extract the
# shortest reads.
# -L , --lgsreads Long-read sequencing data
# -W , --wgs1 Path to WGS reads (read 1)
# -w , --wgs2 Path to WGS reads (read 2)
# -N , --NextPolish Path to NextPolish tool
# -m , --motif Telomeric repeats sequences, e.g., plant: CCCTAAA(TTTAGGG), animal:
# TTAGGG(CCCTAA), etc.
# -M , --motif_num Input the number of bases of the telomere motif
# --Normal Execute the command according to the general process
# --dir_Max Select the telomere reads obtained by polishing the longest reads to
# add to the genome
# --dir_Min Select the telomere reads obtained by polishing the shortest reads
# to add to the genome
# -t , --threads Number of threads to use (default: 20)
$ telocomp_Complement --Normal \
-G geonome.fasta \
--dir_contigs files_NP \
--dir_trim_L trim_L \
--dir_trim_R trim_R \
-L HiFi.fastq.gz \
-W WGS_f1.fq.gz \
-w WGS_r2.fq.gz -t 50 \
-N /PATH/NextPolish \
-m CCCTAAA -M 7
Example
The following pictures show the left and right ends of chromosome 2 of Morus notabilis.Telomere density distribution diagram of chromosome ends with telomere complementation(The following pictures show the left and right ends of chromosome 2).
Telomere density plot at the left end :
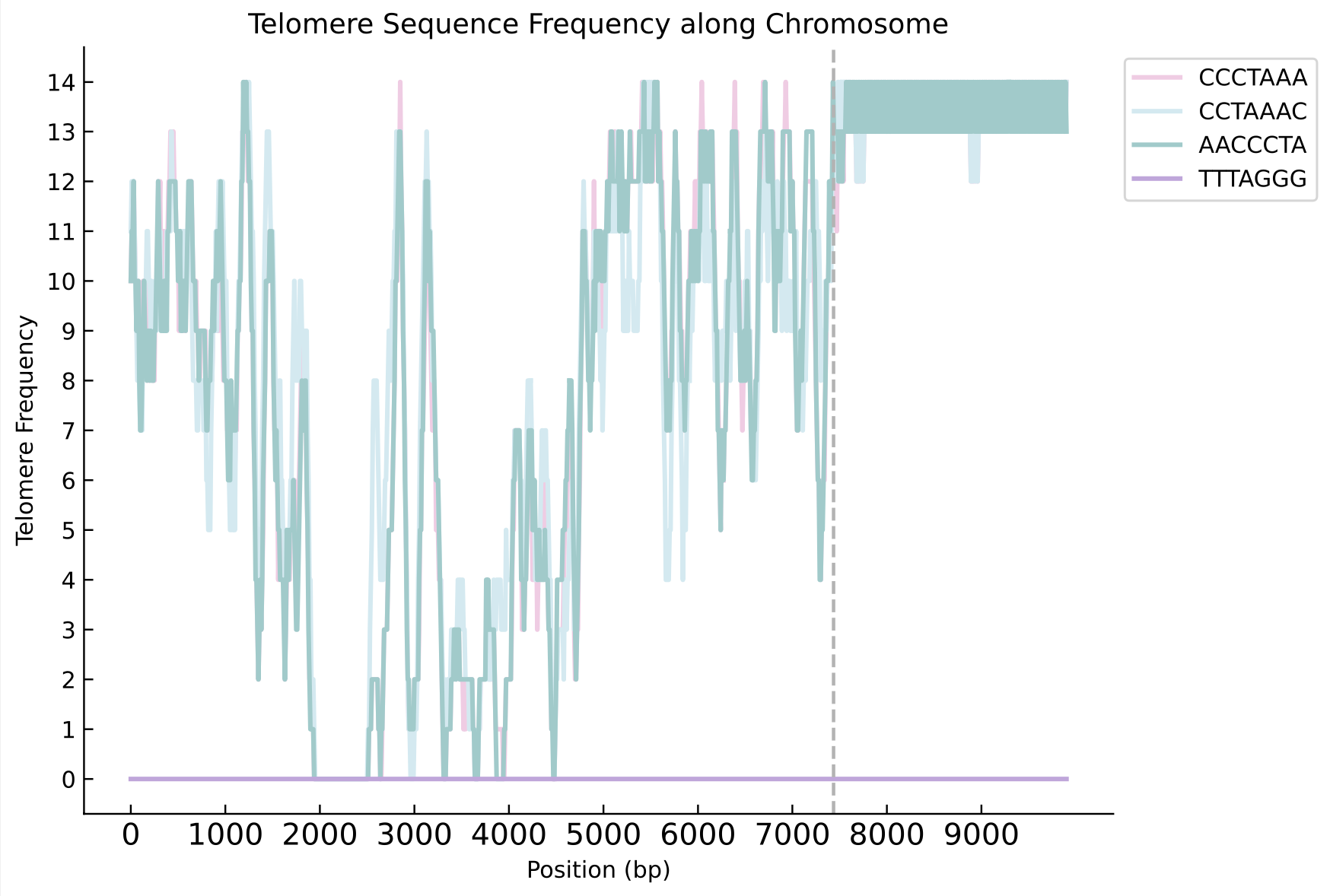
Telomere density plot at the right end: